All About the Turbidity-Based Lysozyme Activity Assay
by Katharine Martin

by Katharine Martin
Lysozyme is an enzyme that breaks down peptidoglycan, and it is often used in protein purification, RNA purification and Northern blot to lyse cells in bacterial expression systems.
When using lysozyme for protein purification, researchers want to ensure good enzymatic activity because it leads to more efficient cell lysis, better yields, and better target protein purity.
One of the ways researchers will assess lysozyme activity is to use a turbidity-based activity assay.
The turbidity-based lysozyme activity assay measures lysozyme enzymatic activity, allowing researchers to evaluate a bacterial cell suspension for a reduction in turbidity (cloudiness). As lysozyme breaks down bacterial cell walls, cell suspension turbidity decreases.
Enzymes speed up reactions and lower the activation energy needed for the reaction to occur. And enzymatic activity refers to an enzyme’s ability to catalyze a reaction. In the case of lysozyme, researchers would measure enzymatic activity to determine its ability to break down peptidoglycan.
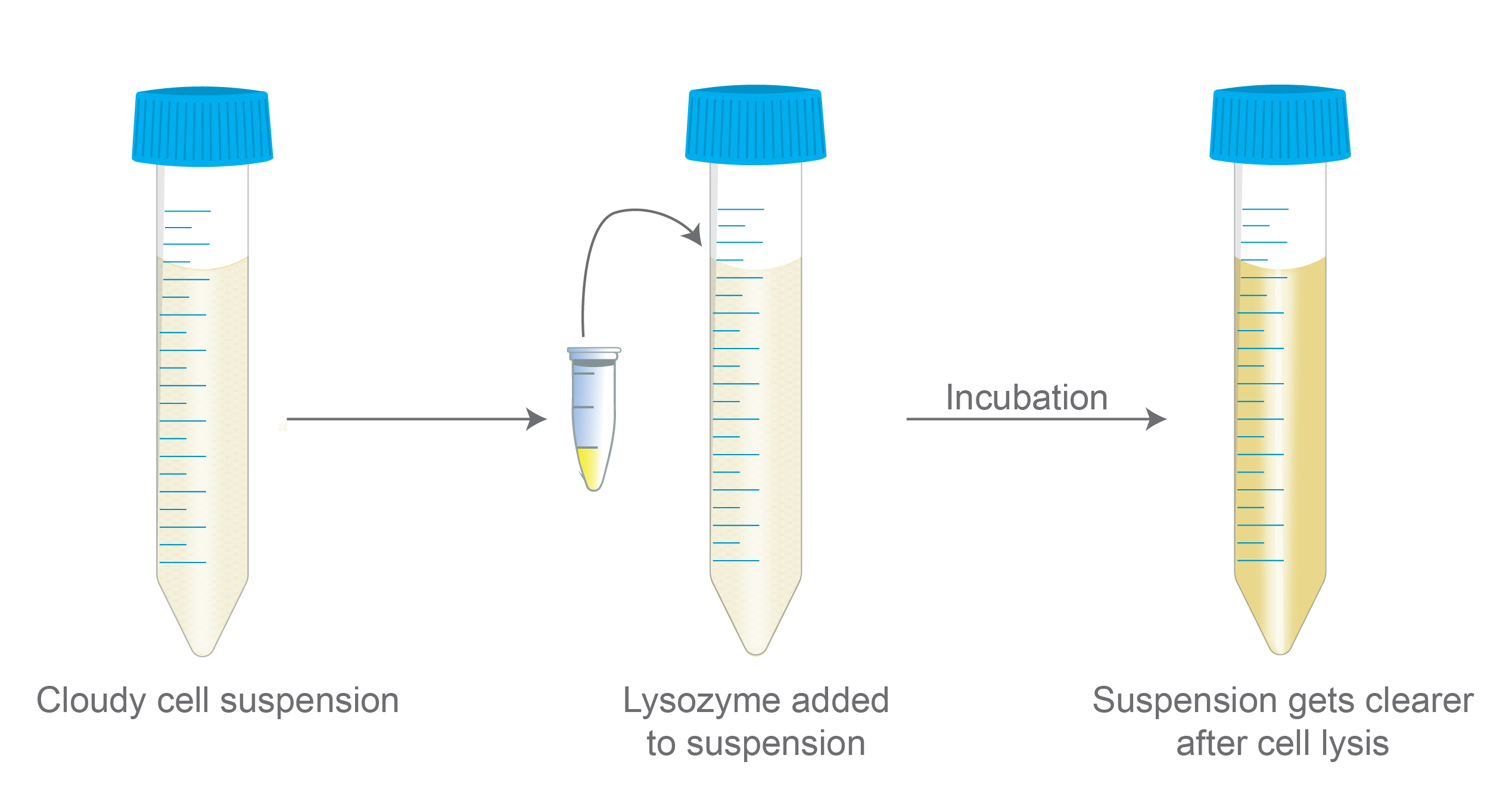
Figure 1. Illustrates the results when lysozyme is
added to a cell suspension. After incubation with lysozyme and cell lysis
occurs, the cell suspension becomes clearer.
During the assay, researchers use a spectrophotometer to measure the optical density of the suspension after lysozyme is added and breaks down the bacterial cell wall.
To understand the principle of the turbidity-based lysozyme activity assay, it’s important to have a general idea of how lysozyme breaks down the cell wall.
The way lysozyme works is by hydrolyzing the glycosidic bonds of the peptidoglycan layer of bacterial cell walls.
The peptidoglycan layer of the cell wall is a structural component, and hydrolysis ultimately leads to bacterial cell lysis.
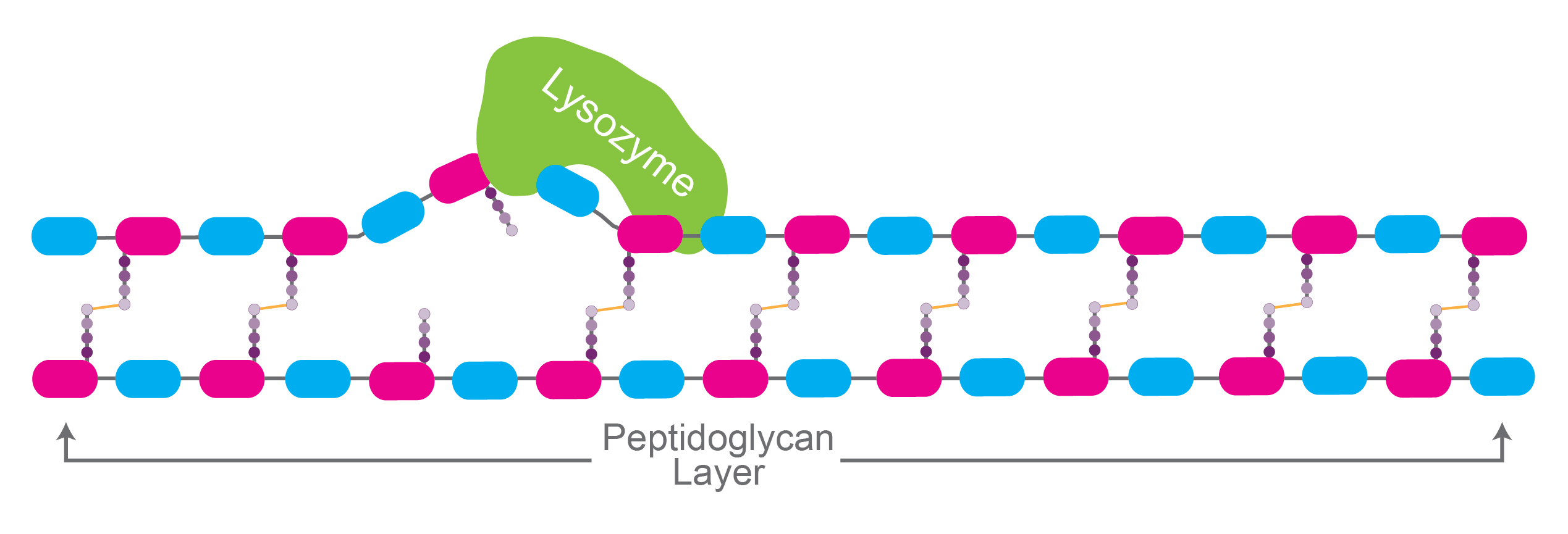
Figure 2. Simplified illustration of lysozyme
hydrolyzing glycosidic bonds in the peptidoglycan layer of the cell wall.
In terms of the mechanism, lysozyme has 2 catalytic sites made up of specific amino acid residues that are needed for the hydrolysis reaction to occur.
When lysozyme binds to the carbohydrate chain of the peptidoglycan, its catalytic sites interact with the 4th and 5th carbohydrate rings. Here, the amino acids Glu35 and Asp52 play crucial roles in the process, and their joint functions lead to lysozyme cleaving the peptidoglycan layer causing cell lysis.
As cell lysis occurs, suspension turbidity (cloudiness) decreases, and this is a key principle behind the assay.
What causes the suspension to actually become less turbid is the cell contents being released into the cell suspension, leading to a decrease in particle size and a reduction in light scattering.
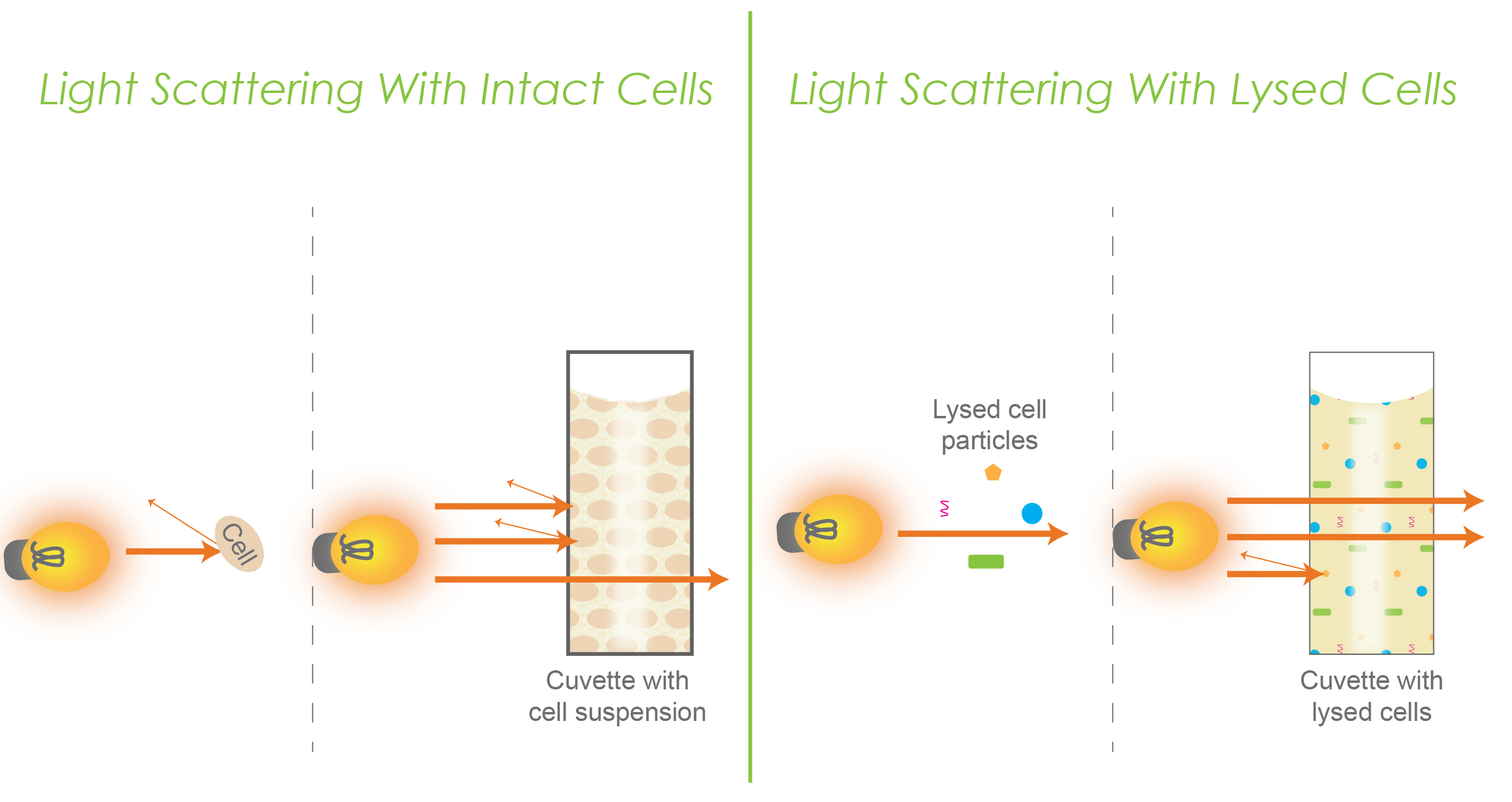
Figure 3. Represents light scattering in intact cells (left) versus lysed cells (right). Each of the two panels have an individual conceptual representation as well as a representation within a cuvette.
Consider the fact that intact bacterial cells are larger in size with a more complex structure that contributes to greater turbidity. This is represented in the left panel of figure 3. Here you can see that as light reaches a cell, some light will bounce back, and in a cuvette, a little bit of light can pass through while some light bounces back and scatters.
When cellular components are released and broken down, less light scattering occurs, which causes a decrease in turbidity. This is shown in the right panel of figure 3.
Researchers generally use a spectrophotometer or microplate reader to measure the optical density (OD) in a lysozyme activity assay.
To begin, a bacterial cell suspension is prepared using the bacterium Micrococcus luteus along with necessary buffers.
Micrococcus luteus is ideal for measuring lysozyme activity because it is gram-positive, which makes it more susceptible to lysozyme, and because it is easy to culture.
Alternatively, E. coli, which is widely used in molecular biology is not suited for this assay because of its gram-negative cell wall. This outer layer makes E. coli more resistant to lysozyme’s lytic action compared to Micrococcus luteus.
The reason for this is because gram-positive microorganisms have a thick layer peptidoglycan in their cell wall. Since peptidoglycan is a substrate for lysozyme, cells of these organisms are susceptible to this enzyme. On the other hand, with only a thin peptidoglycan layer in the cell wall, gram-negative organisms are resistant to lysozyme degradation.
The assay is set up in a cuvette when using a spectrophotometer or in wells when reading a microplate.
Researchers will first take a baseline measurement to measure the initial turbidity without lysozyme. Afterward, lysozyme will be added to a bacterial cell suspension followed by a short incubation period to allow lysozyme to lyse the cells.
Finally, the optical density is measured and recorded.
Lysozyme activity, like any other enzyme’s activity, can vary based on different conditions such as temperature, time, concentration and pH.
In order to understand activity and optimize certain conditions, enzymatic activity is recorded and plotted in an enzyme kinetic curve.
Performing a kinetic curve can give you more insight into the rate of lysozyme-mediated cell lysis and will let you analyze performance over time.
Enzyme kinetic curves let you do the following:
The basic process of conducting a kinetic curve works the same way as the lysozyme activity assay in that you will prepare your bacterial suspension for the assay including a control, and then you would get your baseline readings (no lysozyme).
The next step would be adding your lysozyme. If you’re measuring different concentrations, you would set up your experiment accordingly. If you were examining activity over time, you would incubate the bacterial cells with lysozyme and record light absorption at different time intervals.
Once you have your recordings, you can begin examining your data to determine the change / decrease in turbidity. This is done by subtracting the absorbance at a given time interval from the baseline absorbance.
Something important to note about absorbance is that when it comes to measuring lysozyme activity using M. luteus, the standard practice across a wide range of protocols is to measure absorbance at 450 nm due to sensitivity (in contrast to E. coli and other bacteria that is measured at 600 nm).
Table 1. Example of absorbance recording at different time intervals.

Calculating lysozyme activity depends on your experimental setup and needs. Commonly, researchers calculate the decrease of turbidity over time, or the change in turbidity/time (ΔA/T).
In other cases, activity is stated as the percentage of turbidity decrease. This would be calculated by taking the change in absorbance over the baseline measurement (ΔA/A0).
Your data and calculations can then be represented graphically.
Keep in mind that it is important to consult similar research when planning your approach to the kinetic curve because units, calculations, and recording may vary depending on your experimental requirements.
When examining the activity of lysozyme, the basic procedure involves preparing the bacterial cell suspension, setting up the assay in wells or cuvettes, getting a baseline reading, incubating the cells with lysozyme, measuring turbidity, recording data and then analyzing it.
Typically, a lysozyme-sensitive bacterium called Micrococcus luteus (formerly Micrococcus lysodeikticus) is used for this assay. However, researchers may use other bacteria in order to suit their specific research needs or to study sensitivity in other species.
When preparing your cell suspension, you will want to adjust the concentration to be based on the lysozyme activity you expect as well as the expected bacterial sensitivity to lysozyme.

A His-tag is a stretch of 6-10 histidine amino acids in a row that is used for affinity purification, protein detection, and biochemical assays. His-tags...

Competent cells such as DH5a, DH10B, and BL21 will maintain their transformation efficiency for at least a year with proper storage. It is important to...

Ni2+ ions give nickel agarose beads their characteristic blue color. This blue color can fade or disappear completely when loading his-tagged proteins onto the column....

Nickel agarose beads change from blue to a brown or black color when the nickel ions have been reduced from a Ni2+ to a Ni1+...
