Probing the Depths of Biology: 4 Ways Purified Proteins Drive Research
by Simon Currie, Ph.D.

by Simon Currie, Ph.D.
Our cells are beautifully complex 4D puzzles - consisting of millions of molecular pieces that engage in dynamic interactions to sustain life as we know it. In such an immensely complicated puzzle, how do we know how different individual pieces contribute to the whole?
One way to answer this question is to simplify the system to look at just one, or a few pieces at a time.
In this reductive strategy, scientists purify a protein away from the rest of the cell to see what that protein does on its own. This important information can then be applied back to the native cellular context to help us understand how proteins contribute to the immense puzzles that are whole cells, or even entire organisms.
Another way to examine this question is by observing what pieces are changed in diseases. Observing which proteins are mutated in disease, and then comparing the function of wild type and mutant proteins provides important clues into the molecular basis of a particular disease. This strategy can also provide direction into what actions might help fix an aberrant disease state.
In this article, we will explore how purified proteins are used to learn more about fundamental biology, as well as the diseases that ail us. Four of the main research applications of purified proteins are to investigate:
We can think of a cell like a car in that there are distinct parts that perform different functions. For example, in a car the engine propels the car forward, and the tires contact the road to make this happen. Fortunately for us, a good mechanic knows all the many parts of a car, their functions, and how to fix them when a part goes bad.
Although a car is a useful starting analogy, the reality is a bit more complicated in cells that contain many thousands of different types of parts, most of which we don’t even know their functions.
Also, the interactions in cells are dynamic and change over time. In a car this would be as if the steering wheel controlled the steering while the car was running, but then turned into an engine cleaning device while the car is off.
Among all of this molecular and temporal complexity, how do we actually know what any individual proteins do?
Historically, scientists figured this out by purifying a protein away from the rest of the cell and testing their properties in isolation (Figure 1). Such studies are called in vitro studies, Latin for “within the glass,” as in within a glass test tube or flask.
These types of studies revealed fundamental biological principles including understanding how:

Figure 1. A
pink triangle protein converts a purple circle into a green circle. Scientists
purify this protein from cells (left) to carefully examine and understand this
activity
in vitro (right).
These types of studies help us to understand life at the cellular and molecular level. Additionally, these basic science studies form the foundation for understanding what goes wrong in a disease, and how we can fix it.
Think of it this way – if your mechanic has never heard of brakes and doesn’t know what they do, you’re probably not going to get your brakes fixed!
One way to figure out what a protein does is to see what it looks like. Scientists use structural studies to get a picture of how proteins work. These studies are also very useful in determining how protein structure and function are altered in diseases.
X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy (cryo-EM) are common techniques for determining a protein’s structure. These techniques usually require purifying the protein of interest to near homogeneity to obtain high-quality structures.
In x-ray crystallography, crystals are formed using purified proteins, which are then blasted with x-rays (Figure 2). The diffraction pattern of the x-rays off the protein crystal is used to figure out the structure of the protein within the crystal.
The strength of x-ray crystallography is that it provides very high-resolution structures of proteins, and other biomacromolecules like RNA and DNA.
Proteins are made up of amino acids, which themselves are made up of carbon, oxygen, nitrogen, and sulfur atoms. Good crystal structures allow us to know the location of these atoms at angstrom resolution. An angstrom is 10-10 M; smaller than one billionth of a meter!
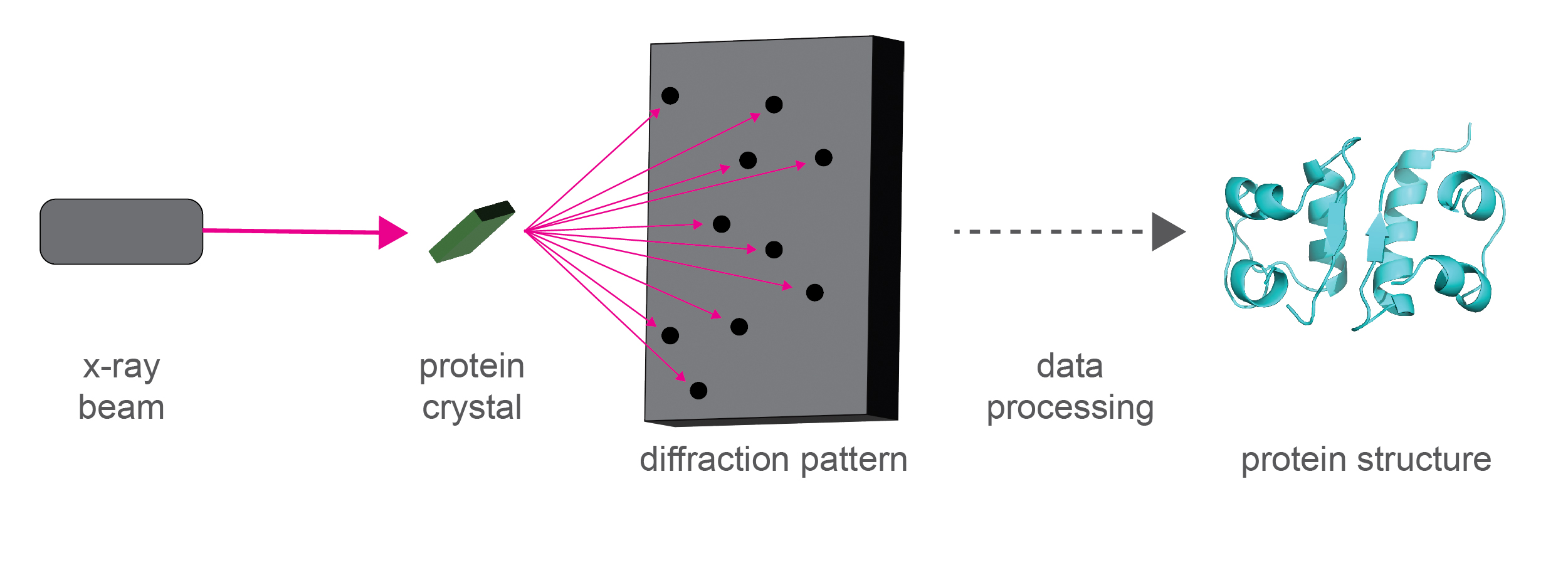
Figure 2. Diagram of how x-ray crystallography works.
Purified proteins are a powerful tool for learning more about fundamental biology, and the diseases that ail us. They are also critical reagents in the quest to treat and cure diseases.
The diffraction pattern of x-rays scattering off a protein crystal are used to solve the structure of the protein in the crystal.
NMR spectroscopy can also solve protein structures, though not to as high of resolution as x-ray crystallography. Not every protein forms crystals, so NMR is a tractable route for understanding the structure of such proteins.
Moreover, the real strength of NMR is in understanding dynamic information about protein structures, while x-ray crystallography is like capturing a snapshot. Think of a gymnast performing the vault routine. If you only had a snapshot of their start and of their landing you would have missed all of the exciting moves they performed while up in the air! NMR spectroscopy provides a lot of important structural information about the motions of proteins and can even reveal “invisible” protein states that are important for biological function, but not captured by x-ray crystallography.
X-ray crystallography and NMR spectroscopy have been used to study protein structure for many decades. In contrast, the popularity of cryo-EM for this purpose has historically been low but has risen drastically in the last 10 years.
This rise in usage correlates with a technological resolution revolution that enables cryo-EM to now provide fairly high-resolution protein structures. However, in most cases not as high of resolution as x-ray crystallography, yet.
Cryo-EM also provides dynamic information about the different conformations that a protein can take. Also, cryo-EM is particularly well suited at studying a big protein and protein complexes that are difficult to study by x-ray crystallography and NMR.
The differences in these methods mean that distinct purification strategies may be used for proteins depending on which method will be used.
X-ray crystallography requires very pure protein in a single conformation, and therefore multiple purification steps are used. In contrast, it is informative to have multiple conformations in cryo-EM, so purification steps that separate different conformations are skipped for this purpose.
A requirement, and limitation, of these methods is purifying proteins away from their cellular environment to solve their structure. Scientists perform careful functional and cellular studies to check whether what they are learning about proteins in isolation is relevant to the protein’s structure and function in a cell.
However, a related technique, cryo-electron tomography (cryo-ET), may be able to solve the structures of proteins in cells without the need for purification.
A recent study used cryo-ET to solve the structure of microtubules in mouse sperm cells (Chen et al., 2023).
However, microtubules have a very particular shape, and are relatively easy to identify in cells by cryo-ET. The especially exciting part of this study is that they were able to identify novel proteins that interact with microtubules by docking the AlphaFold predicted structures of the mouse proteome into the extra bits of their structure that microtubules did not fill out.
So, this approach holds great promise for revealing cellular structures and learning novel biology without requiring prior purification of proteins.
Sickle cell anemia is an inherited disorder characterized by red blood cells that are misshapen and have a reduced capacity to transport oxygen throughout the body.
Hemoglobin is the protein in red blood cells that transports oxygen throughout our bodies. Normally, hemoglobin is present as a tetramer; four hemoglobin proteins interact to execute its oxygen-carrying function (Figure 3). In sickle cell anemia, a mutation in hemoglobin causes it to clump together, or aggregate, with other hemoglobin tetramers (Nishio et al., 1983).
In fact, these aggregates grow so long that they perturb the cell membrane, thereby causing the dysmorphic sickle cell shape of red blood cells. Sickle-shaped cells are sticky and can slow down blood flow, and they easily break open and die. This mutation also reduces the ability of hemoglobin to bind and transport oxygen at the molecular level.
The reduced blood flow, reduced number of live red blood cells, and reduced capacity of hemoglobin to carry oxygen together leads to the anemic symptoms characteristic of sickle cell anemia.

Figure 3. Wild
type hemoglobin forms a tetramer (left and center). Mutations result in
aggregation of hemoglobin tetramers causing sickle cell anemia.
Other protein mutations that cause disease include the following examples. Mutations in dystrophin, huntingtin, amyloid beta, and prion protein cause these proteins to aggregate in Duchenne muscular dystrophy, Huntington’s disease, Alzheimer’s disease, and mad cow disease, respectively. Mutations in CFTR deactivate this protein’s function leading to cystic fibrosis. The aberrant fusion of BCR and ABL proteins into a single protein, BCR-ABL, hyperactivates this protein leading to chronic myeloid leukemia. Mutations in androgen receptor leads to its overexpression and drive prostate cancer. Conversely, mutations that destabilize BRCA2 result in breast cancer.
Genetic studies often point out which genes and proteins are mutated in particular diseases. However, as these examples illustrate, disease can arise from any number of things going wrong with crucial proteins – overexpression or underexpression, hyperactivation or deactivation of protein function, or aberrant aggregation.
This is why purifying mutant forms, and comparing them to the wild type protein, can help reveal how a mutation affects a protein, and what might be done to fix the deleterious impact.
In diseases such as those discussed here, a single protein is driving much of the disease pathology, making it an important potential drug target.
Purified proteins are key reagents that help scientists discover and design new drugs to treat disease. Scientists use purified proteins to develop assays that measure key protein functions and use these assays to find new inhibitors of protein function.
For example, in the case of hyperactive BCR-ABL, we would want to find an inhibitor that reduces protein activity. In the case of aggregating amyloid beta in Alzheimer’s, we would want to find a method to prevent the protein from aggregating and/or to get rid of aggregated protein.
Knowing a protein’s structure can also help scientists rationally design new drugs that fit into and block functionally-important sites on a protein, like a key into a lock (Figure 4).

Figure 4.
Structure of chronic myeloid leukemia drug imatinib (orange) bound to BCR-ABL (gray,
PDB: 2HYY, Cowan-Jacob et al., 2007).
In some cases, these strategies are incredibly successful!
For example, new drugs for chronic myeloid leukemia and prostate cancer have been discovered that drastically improve patient survival and quality of life compared to previous treatments.
On the other hand, strategies to disrupt amyloid-beta aggregation in Alzheimer’s disease so far have been unsuccessful for treating patients. What is the problem here? We don’t conclusively know yet, but it could be that:
While these strategies don’t end up being successful in every case, the use of purified proteins to better understand and treat diseases has had a major impact on numerous patients!
Now that we’ve covered a few of the key applications of purified proteins, you can see why they are such important research tools that help us understand fundamental biology, many diseases, and how to correct disease states. If the disease-related aspects of this article piqued your interest, visit our related article on the medical applications of purified proteins.
Baldwin, A. J., & Kay, L. E. (2009). NMR spectroscopy brings invisible protein states into focus. Nature chemical biology, 5(11), 808–814. https://doi.org/10.1038/nchembio.238
Bäuerlein, F. J. B., & Baumeister, W. (2021). Towards Visual Proteomics at High Resolution. Journal of molecular biology, 433(20), 167187. https://doi.org/10.1016/j.jmb.2021.167187
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., Shindyalov, I. N., & Bourne, P. E. (2000). The Protein Data Bank. Nucleic acids research, 28(1), 235–242. https://doi.org/10.1093/nar/28.1.235
Berman, H., Henrick, K., & Nakamura, H. (2003). Announcing the worldwide Protein Data Bank. Nature structural biology, 10(12), 980. https://doi.org/10.1038/nsb1203-980
Campbell, I. D., Dobson, C. M., & Williams, R. J. (1985). The study of conformational states of proteins by nuclear magnetic resonance. The Biochemical journal, 231(1), 1–10. https://doi.org/10.1042/bj2310001
Cerione R. A. (1991). Reconstitution of receptor/GTP-binding protein interactions. Biochimica et biophysica acta, 1071(4), 473–501. https://doi.org/10.1016/0304-4157(91)90007-j
Chen, C. D., Welsbie, D. S., Tran, C., Baek, S. H., Chen, R., Vessella, R., . . . Sawyers, C. L. (2004). Molecular determinants of resistance to antiandrogen therapy. Nat Med, 10(1), 33-39. doi:10.1038/nm972
Chen, Z., Shiozaki, M., Haas, K. M., Skinner, W. M., Zhao, S., Guo, C., Polacco, B. J., Yu, Z., Krogan, N. J., Lishko, P. V., Kaake, R. M., Vale, R. D., & Agard, D. A. (2023). De novo protein identification in mammalian sperm using in situ cryoelectron tomography and AlphaFold2 docking. Cell, S0092-8674(23)01038-3. Advance online publication. https://doi.org/10.1016/j.cell.2023.09.017
Cleton-Jansen, A. M., Collins, N., Lakhani, S. R., Weissenbach, J., Devilee, P., Cornelisse, C. J., & Stratton, M. R. (1995). Loss of heterozygosity in sporadic breast tumours at the BRCA2 locus on chromosome 13q12-q13. British journal of cancer, 72(5), 1241–1244. https://doi.org/10.1038/bjc.1995.493
Collins, S. J., & Groudine, M. T. (1987). Chronic myelogenous leukemia: amplification of a rearranged c-abl oncogene in both chronic phase and blast crisis. Blood, 69(3), 893-898. doi:10.1182/blood.V69.3.893.893
Cowan-Jacob, S. W., Fendrich, G., Floersheimer, A., Furet, P., Liebetanz, J., Rummel, G., Rheinberger, P., Centeleghe, M., Fabbro, D., & Manley, P. W. (2007). Structural biology contributions to the discovery of drugs to treat chronic myelogenous leukaemia. Acta crystallographica. Section D, Biological crystallography, 63(Pt 1), 80–93. https://doi.org/10.1107/S0907444906047287
Deininger, M. W. N., Goldman, J. M., Lydon, N., & Melo, J. V. (1997). The Tyrosine Kinase Inhibitor CGP57148B Selectively Inhibits the Growth of BCR-ABL–Positive Cells. Blood, 90(9), 3691-3698. doi:10.1182/
Fedele E. (2023). Anti-Amyloid Therapies for Alzheimer's Disease and the Amyloid Cascade Hypothesis. International journal of molecular sciences, 24(19), 14499. https://doi.org/10.3390/ijms241914499
Hill, A. F., Desbruslais, M., Joiner, S., Sidle, K. C., Gowland, I., Collinge, J., Doey, L. J., & Lantos, P. (1997). The same prion strain causes vCJD and BSE. Nature, 389(6650), 448–526. https://doi.org/10.1038/38925
Ho, B., Baryshnikova, A., & Brown, G. W. (2018). Unification of Protein Abundance Datasets Yields a Quantitative Saccharomyces cerevisiae Proteome. Cell systems, 6(2), 192–205.e3. https://doi.org/10.1016/j.cels.2017.12.004
Iannuzzi, M. C., Stern, R. C., Collins, F. S., Hon, C. T., Hidaka, N., Strong, T., Becker, L., Drumm, M. L., White, M. B., & Gerrard, B. (1991). Two frameshift mutations in the cystic fibrosis gene. American journal of human genetics, 48(2), 227–231.
Kao, L., Krstenansky, J., Mendell, J., Rammohan, K. W., & Gruenstein, E. (1988). Immunological identification of a high molecular weight protein as a candidate for the product of the Duchenne muscular dystrophy gene. Proceedings of the National Academy of Sciences of the United States of America, 85(12), 4491–4495. https://doi.org/10.1073/pnas.85.12.4491
Murphy, M. P., & LeVine, H., 3rd. (2010). Alzheimer's disease and the amyloid-beta peptide. J Alzheimers Dis, 19(1), 311-323. doi:10.3233/JAD-2010-1221
Nierhaus, K. H., & Dohme, F. (1974). Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America, 71(12), 4713–4717. https://doi.org/10.1073/pnas.71.12.4713
Nishio, I., Tanaka, T., Sun, S. T., Imanishi, Y., & Ohnishi, S. T. (1983). Hemoglobin aggregation in single red blood cells of sickle cell anemia. Science (New York, N.Y.), 220(4602), 1173–1175. https://doi.org/10.1126/science.6857241
Pauling, l., & Itano, H. A. (1949). Sickle cell anemia a molecular disease. Science (New York, N.Y.), 110(2865), 543–548. https://doi.org/10.1126/science.110.2865.543
Persichetti, F., Ambrose, C. M., Ge, P., McNeil, S. M., Srinidhi, J., Anderson, M. A., Jenkins, B., Barnes, G. T., Duyao, M. P., & Kanaley, L. (1995). Normal and expanded Huntington's disease gene alleles produce distinguishable proteins due to translation across the CAG repeat. Molecular medicine (Cambridge, Mass.), 1(4), 374–383.
Rizo J. (2018). Mechanism of neurotransmitter release coming into focus. Protein science : a publication of the Protein Society, 27(8), 1364–1391. https://doi.org/10.1002/pro.3445
Schubert, T., & Römer, W. (2015). How synthetic membrane systems contribute to the understanding of lipid-driven endocytosis. Biochimica et biophysica acta, 1853(11 Pt B), 2992–3005. https://doi.org/10.1016/j.bbamcr.2015.07.014
Smyth, M. S., & Martin, J. H. (2000). x ray crystallography. Molecular pathology : MP, 53(1), 8–14. https://doi.org/10.1136/mp.53.1.8
Spahn, C. M., & Penczek, P. A. (2009). Exploring conformational modes of macromolecular assemblies by multiparticle cryo-EM. Current opinion in structural biology, 19(5), 623–631. https://doi.org/10.1016/j.sbi.2009.08.001
The PyMOL Molecular Graphics System, Version 2.5.2 Schrödinger, LLC.
Tran, C., Ouk, S., Clegg, N. J., Chen, Y., Watson, P. A., Arora, V., . . . Sawyers, C. L. (2009). Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science, 324(5928), 787-790. doi:10.1126/science.1168175
Traub, P., & Nomura, M. (1968). Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proceedings of the National Academy of Sciences of the United States of America, 59(3), 777–784. https://doi.org/10.1073/pnas.59.3.777
Weir, J. R., Faesen, A. C., Klare, K., Petrovic, A., Basilico, F., Fischböck, J., Pentakota, S., Keller, J., Pesenti, M. E., Pan, D., Vogt, D., Wohlgemuth, S., Herzog, F., & Musacchio, A. (2016). Insights from biochemical reconstitution into the architecture of human kinetochores. Nature, 537(7619), 249–253. https://doi.org/10.1038/nature19333
Williams R. J. (1989). NMR studies of mobility within protein structure. European journal of biochemistry, 183(3), 479–497. https://doi.org/10.1111/j.1432-1033.1989.tb21076.x

Antibiotics and cell selection agents are used to select for specific cell populations to, for instance, generate a stable cell line or conduct a genetic...

Antibiotics are powerful tools that protect cell culture from contamination and help select for transfected cells. However, antibiotics can also have subtle impacts on resistant...

Antibiotics and cell selection agents are used to isolate cells that contain a particular resistance marker from a mixed population. These powerful reagents are used...

During my undergraduate internship I was making different formulations of insulin nanoparticles that would, in theory, be delivered through an inhaler and into the lungs....